
")
Rare Genetic Disorder Treated in the Womb for The First Time

Will SMA disease be cured in the womb, are we moving towards a future where diseases can be prevented in the in-utero period with gene therapy?
Spinal Muscular Atrophy (SMA) is a genetic neuromuscular disorder caused by the degeneration of motor neurons in the spinal cord as a deficiency of the SMN (surviving motor neuron) protein, which leads to muscular atrophy. In this study, Risdiplam, a small molecule drug that improves disease symptoms in patients with SMA by increasing the level of SMN protein, was applied to a fetus with SMA diagnosed by amniocentesis in the intrauterine period. The drug was taken daily for six weeks from 32 weeks of gestation. The girl is now 2 and a half years old and has continued to take the medication since birth. It is reported that no features of SMA, such as hypotonia, weakness, areflexia, etc. have appeared to date. This study demonstrates that the drug, which was applied to only 1 person but had significant effects, may become a preferred treatment method in the treatment of SMA in the future.
Health Outcomes and Drug Utilisation in Children with Noonan Syndrome: A European Cohort Study

Noonan Syndrome in European children, what happens in a decade?
Noonan Syndrome (NS) is an autosomal dominant multisystem disorder. It is the most common RASopathy and is caused by numerous genes encoding various components and regulators of the RAS/MAPK pathway. This multicenter & population-based study analyzed survival rates, hospitalizations, and medication use among children with NS across eleven regions in seven European countries. The findings showed a survival rate of 95% at one year and 92% at ten years, indicating that prognosis improves significantly after the first year of life. Hospitalization was common, with nearly 90% of children admitted in the first year and frequent stays continuing beyond infancy. More than half underwent at least one surgical procedure by age five, and antibiotic use increased noticeably over time. Unlike previous studies that focused on patients referred to tertiary care centers, this study’s strength lies in its population-based approach, including all children diagnosed with NS within the covered regions.
Evaluation of Lyso-Gb1 as a Biomarker for Gaucher Disease Treatment Outcomes Using Data from the Gaucher Outcome Survey
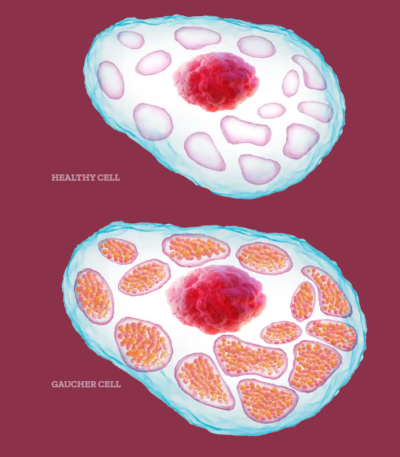
An old biomarker of a rare disease just solidified its place as a valuable diagnostic and monitoring tool in the literature with real-life data!
Gaucher disease is a genetic disorder caused by a deficiency in the glucocerebrosidase (GCase) enzyme, leading to the accumulation of glucocerebroside in the liver, spleen, and bone marrow. Lyso-Gb1 (glucosylsphingosine) is a toxic deacetylated form of glucocerebroside that accumulates in patient plasma, correlating with disease severity and treatment response. It has been a well-established biomarker for Gaucher disease, but this study is the first to leverage real-world data and long-term monitoring to assess its effectiveness in differentiating outcomes between enzyme replacement therapy and treatment-naive patients, supporting its use for personalized disease management.
A Novel Deep Learning Approach for Analyzing Glomerular Basement Membrane Lesions in a Mouse Model of X-Linked Alport Syndrome
A research for GBM in XLAS, explored from multiple perspectives, introduces a novel deep learning-based analysis. Could this pave the way for a new era in pathological assessment?
The study presents a novel deep learning-based approach for detecting and quantifying glomerular basement membrane (GBM) lesions in a mouse model of X-linked Alport syndrome (XLAS) using electron microscopy images. The results demonstrate that the model effectively identifies GBM abnormalities, including thinning, thickening, and splitting, with high accuracy and reproducibility, outperforming traditional manual assessments. This is the first application of deep learning to automate GBM pathology analysis, offering a faster, objective, and quantitative method for evaluating disease progression. The significance of this work lies in its potential to enhance research on Alport syndrome, facilitate drug development, and improve diagnostic precision in glomerular diseases, paving the way for future clinical applications in human patients.
Long-Term Safety and Efficacy of Lenabasum, a Cannabinoid Receptor Type 2 Agonist, in Patients with Dermatomyositis with Refractory Skin Disease: Follow-Up Data from a 3-Year Open-Label Extension Study

Trying to deal with the relentless skin symptoms of dermatomyositis? What if an alternative pathway—one rooted in the body’s endocannabinoid system—could provide lasting relief?
Dermatomyositis (DM) is a rare autoimmune disease characterized by muscle weakness and distinctive skin rashes. A significant subset of DM patients experiences refractory skin disease, where cutaneous manifestations persist despite standard treatments; approximately 25% of DM patients do not respond to corticosteroid therapy, and 25-50% develop adverse reactions. This recent 3-year OLE study measured the sustained safety and efficacy of a selective cannabinoid receptor type 2 (CB2) agonist, Lenabasum. It involved 20 participants, who received lenabasum bi-daily, which it was observed to have significant improvements after week 68. The study showed that even after the trials were discontinued, the improvements regarding the disease sustained.
Is Transthyretin Amyloid Cardiomyopathy Losing Its Rare Disease Classification?

Was ATTR-CM ever a rare disease, or were we simply unable to diagnose it?
Previously thought to be uncommon, ATTR-CM was often misdiagnosed as general heart failure, especially in elderly individuals, leading to an underestimation of its prevalence. However, advancements in imaging techniques, biomarker analysis, and genetic screening have improved diagnostic accuracy, revealing that many previously undiagnosed cases were overlooked rather than nonexistent. Additionally, as life expectancy rises, more individuals reach the age at which wild-type ATTR (wtATTR) manifests, further contributing to higher case numbers. This study highlights how growing recognition and improved detection are shifting the classification of ATTR-CM from a rare disease to a more common condition.